What is clinical study?
What is a Clinical Trial/Study?
A clinical trial/study refers to a test or study conducted on humans to confirm the pharmacokinetics,
pharmacodynamics, pharmacology, and clinical effects of an investigational drug and understand its adverse reactions
for the purpose of proving the safety and effectiveness of the drug.
In other words, it is the process of proving the efficacy and safety of a newly developed drug. It is a scientific study targeting healthy volunteers or patients. It shall be conducted in accordance with the ethical regulations based on the Declaration of Helsinki, domestic clinical trial management standards, and related domestic and foreign regulations. Clinical trials should be conducted only when it is judged that the benefits to the individual subjects and society can outweigh or justify the risks after sufficiently considering the risks and inconveniences predicted from clinical trials. The rights, safety, and welfare of test subjects are the top priorities.
Although the results of clinical trials cannot always be good, it can be an opportunity for patients with cancer or incurable diseases because they may be exposed to new drugs or treatments in development before the drugs are marketed, which can become a hope for treatment. Moreover, the subjects who participate in clinical trials should be respected because they are dedicated with a noble will to help treat patients like themselves.
Classification of Clinical Trials
-
01
Phase 1 clinical trial
The phase 1 clinical trial is a clinical trial that aims to determine the pharmacokinetics, pharmacological action in the human body, side effects, and the safe dose (tolerance) of a drug after administering the drug to a relatively small number of healthy people (usually between 20 and 80 subjects, sometimes less than 20) based on toxicity, absorption, metabolism, excretion, and pharmacological action data obtained from preclinical animal testing of drug candidates.
-
02
Phase 2 clinical trial
Phase 2 is a step to prove the efficacy and safety of a new drug. It is a test to confirm the pharmacological effect and determine the appropriate dosage or usage. It is usually carried out using a limited number of patients who can be carefully examined. The number of target patients is generally between 100 and 200. However, it may be performed in a much larger number of patients for drugs with a range of applicable diseases, such as antimicrobic drugs.
-
03
Phase 3 clinical trial
Phase 3 is conducted after the effectiveness of the new drug has been identified to some extent. It is the last clinical trial to obtain drug approval. It is a test to compare and evaluate the dose, effect, efficacy, and safety of the drug by setting a control group and a treatment group at the same time. The number of target patients depends on the characteristics of the drug. Generally, it is desirable to set the number of target patients to confirm side effects that appear with a probability of 1/1000.
-
04
Phase 4 clinical trial
This is a test to evaluate the long-term efficacy and safety of new drugs after it is sold and used. It may include post-marketing surveillance to obtain additional information on the frequency of side effects of new drugs, studies to search for special pharmacological effects, large-scale follow-up studies to review the effects of drug use on morbidity or mortality, clinical trials on special patient groups that were not reviewed in pre-marketing clinical trials, and post-marketing clinical studies to explore new indications.
- Goals of each clinical trial stage
-
Phase 1
clinical trialIt is carried out in small numbers (20 to 80 people) to discover side effects, determine a safe dose range, and evaluate the safety of an experimental drug or treatment.
-
Phase 2
clinical trialIt is conducted in large numbers (100 to 300 people) to see if an investigational drug or treatment works and obtain more safety-related data.
-
Phase 3
clinical trialIt is performed to confirm the effectiveness of an investigational drug or treatment, observe side effects, compare it with commonly used treatments, and gather information so that this drug and treatment can be approved for safe use.
-
Phase 4
clinical trialA post-marketing study to obtain additional information, including the optimal use, benefits, and risks of a drug.
- Stages and Procedures of New Drug Development
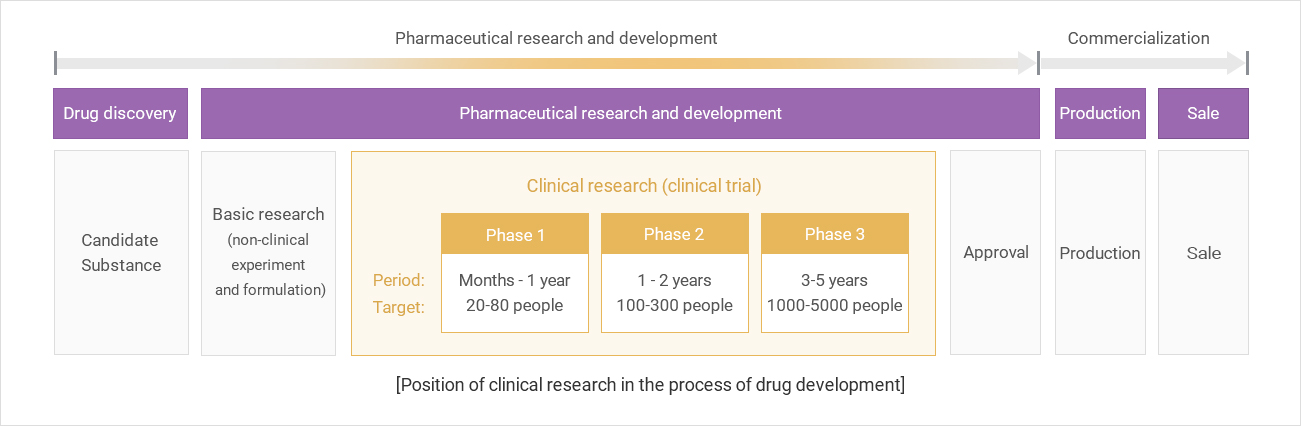
Explanation of terms related to clinical trials
-

01. Clinical Trial/Study
It refers to tests or studies conducted on humans to confirm the pharmacokinetics, pharmacodynamics, pharmacology, and clinical effects of drugs and investigate adverse reactions for the purpose of proving the safety and efficacy of investigational drugs.
-

02. Protocol
It is a document describing the goals, materials and methods, statistical aspects, related organizations, etc. of a clinical trial to provide the background or basis for the clinical trial.
-

03. Clinical Trial/Study
It refers to a procedure that prevents people or departments involved in a clinical trial from knowing about the assigned treatment. Single-blind generally means to blind the subject, while double-blind is blinding the subject, the examiner, the monitor, and, if necessary, the person involved in the data analysis.
-

04. Multicenter Trial
It refers to clinical trials conducted in two or more test institutions according to one clinical trial protocol.
-

05. Serious AE/ADR
Cases falling under any of the following items among adverse reactions or adverse drug reactions that occurred at any dose of a drug used in clinical trials
-

06. Adverse Drug Reaction (ADR)
All adverse and unintended reactions that occurred at any dose of the investigational product. It is not possible to exclude a causal relationship with the investigational product.
-

07. Unexpected Adverse
Drug ReactionWhen it shows differences in the pattern of adverse drug reactions or the degree of harm in light of available drug-related information (e.g., clinical investigator package or attachment of drug products)
-

08. Vulnerable Subject
Subjects whose voluntary participation decisions may be affected due to expectations of benefits associated with participation in clinical trials or concerns about disadvantages they may receive from superiors in the organizational hierarchy if they refuse to participate (e.g., students of medical school, pharmacy school, dental school, nursing school, hospital or research institute employees, pharmaceutical company employees, military personnel, and prisoners), people with incurable diseases, people confined in group facilities stipulated by the Enforcement Regulations, the unemployed, the poor, patients in emergency situations, racial minorities, vagrants, refugees, minors, and subjects unable to give consent of their own free will.
-

09. Institutional Review
BoardA standing committee established independently within the trial institution to protect the rights, safety, and welfare of subjects participating in clinical trials by reviewing and continuously checking the protocol or changed protocol, and methods or provided information to obtain written consent from subjects.
-

10. Impartial Witness
A person who is unrelated to the clinical trial and who can not be unfairly influenced by those involved in the clinical trial. If the subject or the subject's legal representative is illiterate, a person who will be present during the written consent process and will read the written consent form and all written information provided to the subject on behalf of the subject.
-

11. Clinical Trial Pharmacist
A pharmacist who is designated by the head of the testing institution and responsible for the receipt, storage, dispensing, managing, and return of investigational drugs at the test institution.
What Subjects Need to Know Before Deciding to Participate in a Clinical Trial
- The clinical trial is conducted for research purposes.
- Purpose of the clinical trial
- Information on medicines used in the clinical trial and the probability of being randomly assigned to a treatment group or a control group
- Various tests or procedures that subjects will receive during the clinical trial, including invasive procedures
- What the subject must comply with
- Unvalidated experimental aspects of the clinical trial
- If it targets
- Anticipated danger or inconvenience to the subject (it includes the fetus if the subject is a pregnant woman, and it includes the infants if the subject is a breastfeeding woman)
- The fact of an expected benefit to the subject or no expected benefit to the subject through the clinical trial
- Other treatment methods or types that the subject may choose and the potential risks and benefits of these treatment methods.
- If damage due to the clinical trial occurs, the type and amount of compensation or treatments will be given to the subject
- If there is any financial compensation that the subject will receive by participating in the clinical trial, the estimated amount shall be informed. If this amount will be adjusted according to the degree or duration of participation in the clinical trial, it shall be informed as well.
- Expected cost to a subject by participating in a clinical trial
- The subject's decision to participate in the clinical trial must be voluntary. The subject can refuse to participate in the clinical trial or withdraw from participating at any time during a clinical trial without loss of benefits to which he or she may have been entitled.
- The fact that the monitoring personnel, the person conducting the inspection, the review committee, and the Minister of Food and Drug Safety can view the patient's medical records to the extent that the confidentiality of the subject's identity is protected in accordance with the relevant laws and regulations to verify the quality of the clinical trial’s implementation procedures and data. The fact that access to these materials is permitted by a consent signed by the subject or the subject's representative.
- The fact that records that can reveal the subject's identity will be kept confidential and that the subject's identity will be kept confidential when the results of the clinical trial are published.
- The fact that if new information, which may affect the subject's continued participation in the clinical trial, is obtained, the subject or the subject's representative will be notified in a timely manner.
- Whom to contact if the subject needs additional information about the clinical trial and the interests of the subject or in the event of an injury related to the clinical trial.
- When the subject's participation in the clinical trial is suspended and the reason during the clinical trial.
- Expected participation period of the subject in the clinical trial.
- The approximate number of subjects participating in the clinical trial.
Principles of Clinical Trials

Representative Medical Research Ethical Guidelines and Codes
| Name | Year | Announcing Country | Content |
|---|---|---|---|
| The Nuremberg Code | 1947 | Court of Allied Force | Suggested 10 principles to follow when experimenting with human subjects |
| Declaration of Helsinki | 1964 | World Medical Association | Added three principles to the Nuremberg Code
|
| The Belmont Report | 1979 | US | Laid the foundation of future research-related policies by presenting the ethical basic principles of research on human subjects |
| ICH-GCP | 1996 | US, Europe, and Japan | Prepared standardized international guidelines for clinical trials for new drug development |
| KGCP | 2000 | Republic of Korea | Establish guidelines for clinical trials for drug development conducted in South Korea |
| Korean Medical Ethical Guidelines | 2011 | Republic of Korea | Provides basic ethical guidelines for physicians engaged in research |
| Bioethics and Safety Act | 2011 | Republic of Korea | The National Bioethics Committee enacted laws and regulations on institutional bioethics committees, embryo generation and research, and genetic testing research |
Participation and Recruitment Procedures of Clinical Trials
-

NO.1Announcement of applicant recruitment
- If applicants to participate in a clinical trial are needed, the researcher shall post a recruitment announcement or advertisement after receiving approval from the Institutional Review Board (IRB) for the consent statement and recruitment announcement.
- Recruitment announcement or advertisement shall use the recruitment notice section on the website of the clinical trial center, the website of hospitals or related institutions, and/or if necessary, mass media (newspapers or broadcasting).
-

NO.2Application for participation in clinical trials
- Although there are slight differences between the centers or institutions that host clinical trials, they generally accept applications in the following way.
- Online application: Fill out the application form on the website of a clinical trial center (or related institution) - In-person application: Fill out the application form at related institutions and clinical trial centers - Phone (or fax) application: Call the relevant department of a clinical trial center and provide simple information orally
- Even if an applicant cannot participate because there was no suitable trial at the time of application, the applicant may be contacted individually if the participant wishes to participate later.
- Although there are slight differences between the centers or institutions that host clinical trials, they generally accept applications in the following way.
-

NO.3Individual notification to applicants according to the test schedule
- Notify each applicable person by e-mail or phone and arrange a visit.
-

NO.4Explanation and Preparation of Written Consent Forms
- Applicants who are eligible to participate shall visit the clinical trial center at the scheduled visit time, obtain a sufficient explanation of the study from an authoritative person, such as an investigator or research nurse, and provide written consent by filling out a consent form.
- When a research applicant gives written consent, the applicant must be aware of:
- Objectives and method of the clinical trial - Expected efficacy and effects - Side effects and risks - If the applicant is a patient, whether there are other treatment methods for the current disease. - Confirmation that the applicant will not be penalized for not agreeing to participate. - Possibility of withdrawal at any time by free will after the subject has given consent to participate in the trial - Confirmation that the applicant can receive compensation and treatment in case of damage - Guaranteed confidentiality of the identity - Other human rights protection requirements, etc.
-

NO.5Physical examination (Screening test)
- Only those who signed the written consent will undergo a physical examination.
- The screening test is a process to check whether the applicant is suitable for the clinical trial.
- The items of the screening test vary depending on the study, and it takes about 30 minutes. The results of the screening test can be obtained by contacting the relevant institution or clinical trial center.
- The applicant will receive diverse tests including blood/urine tests, body measurements, blood pressure, and pulse as well as a medical examination by interview by a doctor in charge. Depending on the study, more complex tests may be required.
- Precautions for accurate examination
- No food or drink other than drinking water should be consumed for 10 hours prior to the screening test, - Please avoid excessive exercise or drinking from three days prior to the screening test.
- The applicant may need to visit again for a retest depending on the test results.
-

NO.6Selection of suitable subjects and individual notification
- Only those who are suitable according to the results of the physical examination will participate in the clinical trial.
- Results are individually notified after 2 to 4 days (1 week for special tests).
-

NO.7Participation in the main clinical trial
- Subjects who are suitable according to the results of the screening test will participate in the study according to the research plan established by the center.
- Since the clinical trial is conducted according to the study plan as described in the explanation, the subject shall take the clinical trial drug (new drug or control drug) exactly as the researcher directs.
- The trial drug or control drug will be administered once or repeatedly. The subject may receive clinical tests (blood and urine tests) similar to the screening test to confirm safety.
- Blood and urine tests may be performed multiple times to evaluate pharmacokinetics and efficacy. The total amount of blood collected during the period will not exceed the amount of one blood donation.
- Sometimes, hospitalization may be required.
-

NO.8End of clinical trial participation
- If there is no specific finding during the clinical trial process, participation in the trial ends after the planned period.
- If an unusual or severe adverse reaction occurs during the trial, participation in the test may be discontinued based on the judgment of the researcher or the will of the subject.
- If participation terminates due to a side effect caused by the clinical trial, appropriate compensation will be made by the sponsor.